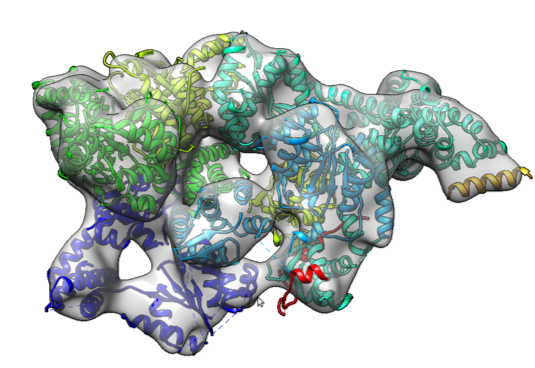
Protein structure determination from low-resolution experimental data
Determining a protein’s three dimensional structure — the position of each of its tens of thousands of atoms — is a key challenge in biochemistry. While many known protein structures were solved using high-resolution techniques, such as X-ray crystallography, for many interesting and biomedically relevant proteins, we can only collect sparse experimental information. That is, we may collect information describing the proteins conformation, but not enough information to uniquely place every atom. Our lab is interested in developing novel computational tools to determine high-resolution structures using only low-resolution data. Key to this is drawing information from the tens of thousands of previously solved structures.
Representative publications:
Cryo-electron microscopy:
- B. Frenz, A. Walls, E. Egelman, D. Veesler, F. DiMaio (2017). A greedy conformational sampling strategy enables automated interpretation of difficult cryoEM maps. Nature Methods 14(8):797-800.
- R. Y.-R. Wang, Y. Song, B. Barad, Y. Cheng, J. Fraser, F. DiMaio (2016). Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife e17219.
- R. Y.-R. Wang, M. Kudryashev, X. Li, E. Egelman, M. Basler, Y. Cheng, D. Baker and F. DiMaio. (2015) Accurate de Novo Protein Structure Determination from Near-atomic resolution Cryo-EM Maps. Nature Methods. 12(4):335-8.
Unphased crystallographic data:
- F. DiMaio, N. Echols, J. Headd, T. Terwilliger, P. Adams, D. Baker (2013). Improved protein crystal structures at low resolution by integrated refinement with Phenix and Rosetta. Nature Methods. 10:1102-4.
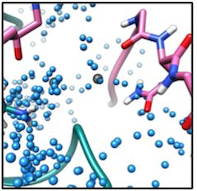
Improved methods for computational modeling of protein conformations
We are interested in developing novel computational methods for modeling the three dimensional structure of proteins. These tools are key to solving structures from low resolution data, predicting structure in the absence of data, and in designing protein sequences to perform novel functions. Advancements in this area of research include improved tools for conformational sampling, better modeling of energetics, and identification and modeling of missing physical parameters in our models. As above, building and validating models against experimental data is key to driving method development forward.
Representative publications:
- R. E. Pavlovicz, H. Park, F. DiMaio (2019). Efficient consideration of coordinated water molecules improves computational protein-protein and protein-ligand docking. bioRxiv 618603; doi: https://doi.org/10.1101/618603
- H. Park, P. Bradley, P. Greisen, Y. Liu, V. Mulligan, D. Kim, D. Baker, F. DiMaio (2016). Simultaneous optimization of biomolecular energy function on features from small molecules and macromolecules. Journal of Chemical Theory and Computation 13;12(12):6201-6212.
- P. Conway, F. DiMaio (2016). Improving hybrid statistical and physical forcefields through local structure enumeration. Protein Sci. 25(8):1525-34.

Prediction and design of symmetric protein assemblies
Symmetry is abundant in nature. Evolution uses symmetry to build large, highly ordered complexes from small individual components. We are interested in developing tools for both prediction and design of symmetrical protein complexes. These tools may allow us to understand the processes that drive symmetric protein assembly, explain why proteins crystalize in the manner that they do (and how to redesign proteins to crystallize in a different way), and develop new protein assemblies with long-range order for materials design.
Representative publications:
- Z. Chen, M.C. Johnson, J. Chen, M.J. Bick, S.E. Boyken, B. Lin, J.J. De Yoreo, J.M. Kollman, D. Baker, F. DiMaio (2019). Self-Assembling 2D Arrays with de Novo Protein Building Blocks. J Am Chem Soc. 141(22):8891-8895.
- S. Gonen, F. DiMaio, T. Gonen, D. Baker (2015). Design of ordered two-dimensional arrays mediated by non-covalent protein-protein interfaces. Science. 348(6241):1365-8.